





Introduction
orfipy is a tool written in python/cython to extract ORFs in an extremely and fast and flexible manner. Other popular ORF searching tools are OrfM and getorf. Compared to OrfM and getorf, orfipy provides the most options to fine tune ORF searches. orfipy uses multiple CPU cores and is particularly faster for data containing multiple smaller fasta sequences such as de-novo transcriptome assemblies. Please read the paper here.
Please cite as: Urminder Singh, Eve Syrkin Wurtele, orfipy: a fast and flexible tool for extracting ORFs, Bioinformatics, 2021;, btab090, https://doi.org/10.1093/bioinformatics/btab090
Installation
Install latest stable version
pip install orfipy
Or install via conda
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
conda create -n orfipy -c bioconda orfipy
Install the development version from source
git clone https://github.com/urmi-21/orfipy.git
cd orfipy
pip install .
or use pip
pip install git+git://github.com/urmi-21/orfipy.git
Examples
Details of orfipy algorithm are in the paper. Please go through the SI if you are interested to know differences between orfipy and other ORF finder tools and how to set orfipy parameters to match the output of other tools.
Below are some usage examples for orfipy
To see full list of options use the command:
orfipy -h
Input
orfipy version 0.0.3 and above, supports sequences in Fasta/Fastq format (orfipy uses pyfastx). Input files can be in .gz format.
Extract ORF sequences and write ORF sequences in orfs.fa file
orfipy input.fasta --dna orfs.fa --min 10 --max 10000 --procs 4 --table 1 --outdir orfs_out
Use standard codon table but use only ATG as start codon
orfipy input.fa.gz --dna orfs.fa --start ATG
Note: Users can also provide their own translation table, as a .json file, to orfipy using --table option. Example of json file containing a valid translation table is here
See available codon tables
orfipy --show-table
Extract ORFs BED file
orfipy input.fasta --bed orfs.bed --min 50 --procs 4
or
orfipy input.fasta --min 50 --procs 4 > orfs.bed
Extract ORFs BED12 file
Note: Add --include-stop for orfipy output to be consistent with Transdecoder.Predict output .bed file.
orfipy testseq.fa --min 100 --bed12 of.bed --partial-5 --partial-3 --include-stop
Extract ORFs peptide sequences using default translation table
orfipy input.fasta --pep orfs_peptides.fa --min 50 --procs 4
API
Users can directly import the ORF search algorithm, written in cython, in their python ecosystem.
>>> import orfipy_core
>>> seq='ATGCATGACTAGCATCAGCATCAGCAT'
>>> for start,stop,strand,description in orfipy_core.orfs(seq,minlen=3,maxlen=1000):
... print(start,stop,strand,description)
...
0 9 + ID=Seq_ORF.1;ORF_type=complete;ORF_len=9;ORF_frame=1;Start:ATG;Stop:TAG
orfipy_core.orfs function can take following arguments
- seq: Required input sequence (str)
- name ['Seq'] Name (str)
- minlen [0] min length (int)
- maxlen [1000000] max length (int)
- strand ['b'] Strand to use, (b)oth, (f)wd or (r)ev (char)
- starts [['TTG','CTG','ATG']] Start codons to use (list)
- stops=['TAA','TAG','TGA'] Stop codons to use (list)
- include_stop [False] Include stop codon in ORF (bool)
- partial3 [False] Report ORFs without a stop (bool)
- partial5 [False] Report ORFs without a start (bool)
- between_stops [False] Report ORFs defined as between stops (bool)
Comparison with getorf and OrfM
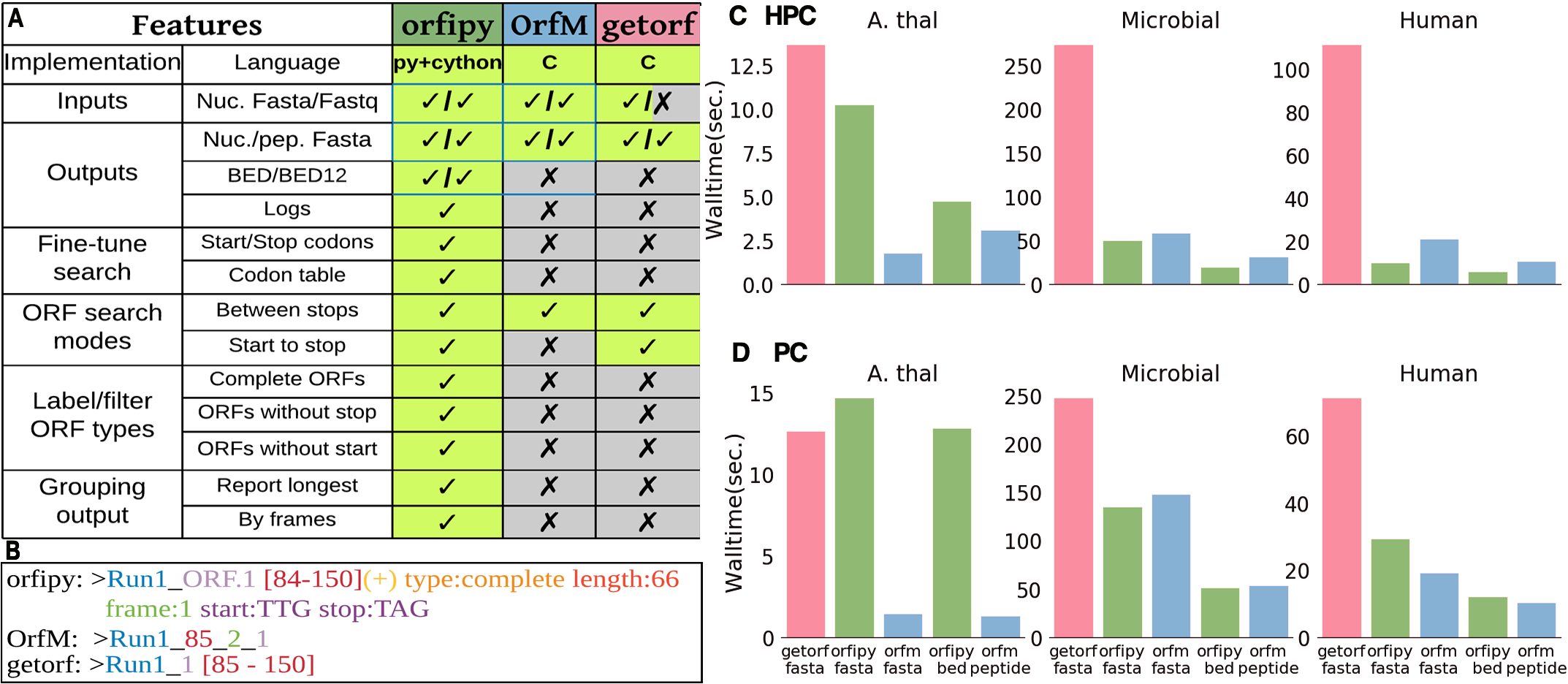 Comparison of orfipy features and performance with getorf and OrfM. Tools were run on different data and ORFs were output to both nucleotide and peptide Fasta files (fasta), only peptide Fasta (peptide) and BED (bed). For details see the publication and SI
Comparison of orfipy features and performance with getorf and OrfM. Tools were run on different data and ORFs were output to both nucleotide and peptide Fasta files (fasta), only peptide Fasta (peptide) and BED (bed). For details see the publication and SI
- orfipy is most flexible, particularly faster for data containing multiple smaller fasta sequences such as de-novo transcriptome assemblies or collection of microbial genomes.
- OrfM is fast (faster for Fastq), uses less memory, but ORF search options are limited
- getorf is memory efficient but slower, no Fastq support. Provides some flexibility in ORF searches.
Funding
This work is funded in part by the National Science Foundation award IOS 1546858, "Orphan Genes: An Untapped Genetic Reservoir of Novel Traits". This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1548562 (Bridges HPC environment through allocations TG-MCB190098 and TG-MCB200123 awarded from XSEDE and HPC Consortium).















 92 Dec 29, 2022
92 Dec 29, 2022
 3 Feb 21, 2022
3 Feb 21, 2022
 20 Nov 24, 2022
20 Nov 24, 2022
 177 Dec 22, 2022
177 Dec 22, 2022
 17 Dec 11, 2022
17 Dec 11, 2022
 1 Feb 23, 2022
1 Feb 23, 2022
 14 Aug 17, 2022
14 Aug 17, 2022
 2 Dec 16, 2021
2 Dec 16, 2021
 37 Dec 06, 2022
37 Dec 06, 2022
 8.3k Jan 08, 2023
8.3k Jan 08, 2023
 2 Jan 01, 2022
2 Jan 01, 2022
 62 Dec 23, 2022
62 Dec 23, 2022
 26 Jan 01, 2023
26 Jan 01, 2023
 170 Dec 11, 2022
170 Dec 11, 2022
 2 Jan 11, 2022
2 Jan 11, 2022
 16 Nov 15, 2022
16 Nov 15, 2022
 8 Jul 05, 2022
8 Jul 05, 2022
 24 Dec 19, 2022
24 Dec 19, 2022
 129 Dec 21, 2022
129 Dec 21, 2022
 4 Oct 24, 2022
4 Oct 24, 2022